
Maladie de Vaquez: la saignée est dépassée?
Dans le traitement de la maladie de Vaquez (MV), la saignée continue d’être une option thérapeutique de première ligne, mais elle entraîne à long terme une carence en fer. Les nouvelles thérapies comme l’inhibition des JAK1/2 avec le ruxolitinib ou le ropeginterféron alfa-2b constituent une avancée majeure dans le traitement à long terme de la MV, car elles permettent de réduire en continu la charge allélique et de prolonger la survie sans événement.
Keypoints
-
Le traitement par saignée seul, en association avec l’AAS, n’influence pas la progression de la maladie vers la myélofibrose ou la LMA.
-
Le rôle de la saignée est de permettre un contrôle initial rapide de l’hématocrite afin d’éviter les événements thromboemboliques aigus.
-
Un traitement par saignées à long terme entraîne des symptômes de carence en fer avec une réduction de la qualité de vie.
-
Le potentiel de modification de la maladie de l’interféron alfa a été démontré tant chez les patient·es à haut risque que chez les patient·es à faible risque.
Depuis la première description de la maladie de Vaquez (MV) en 1892, la saignée est traditionnellement utilisée comme option thérapeutique. Aujourd’hui encore, la saignée est utilisée comme traitement de première ligne relativement rapide chez la majorité des patient·es atteint·es de MV récemment diagnostiquée dans le but de prévenir les complications aiguës comme l’infarctus du myocarde ou l’accident vasculaire cérébral. Les saignées répétées permettent de maintenir l’hématocrite en dessous de la valeur cible définie de 45% chez une partie des patient·es, sans toutefois intervenir dans la pathophysiologie complexe de la maladie. Les symptômes de carence en fer qui s’installent constituent un inconvénient de la thérapie par saignée à long terme.
Réponse moléculaire et EFS prolongée avec l’inhibition des JAK
La mutation V617F de JAK2, décrite pour la première fois en 2005 dans les néoplasies myéloprolifératives (NMP), est détectable chez plus de 95% des patient·es atteint·es de MV et entraîne une activation constitutionnelle de la voie de signalisation JAK-STAT et une hyperprolifération hématopoïétique.1 Les objectifs du traitement sont, outre la prévention des complications thromboemboliques, l’amélioration de la qualité de vie et surtout une modification de l’évolution de la maladie.
Le ruxolitinib, un inhibiteur de JAK1/2, a été comparé à la BAT («best available therapy») dans la maladie de Vaquez dans le cadre des études randomisées de phaseII MAJIC-PV2 et de phaseIIIb RESPONSE-2.3,19 Le ruxolitinib entraîne un contrôle hématologique amélioré et durable (RC selon les critères ELN4), une maîtrise efficace des symptômes et une amélioration de la qualité de vie. Par rapport à la BAT, seule la moitié environ des patient·es du bras ruxolitinib ont eu besoin d’au moins une saignée supplémentaire pour contrôler leur hématocrite (objectif ≤45%).2
Lors du suivi à 5ans de l’étudeMAJIC-PV, en 2023, il a été démontré que la survie sans événement (EFS) était significativement prolongée avec le ruxolitinib. De plus, une réponse moléculaire est obtenue: diminution de la charge allélique JAK2 («variant allele frequency», VAF) dans le sang périphérique. Le délai médian avant l’obtention d’une réponse moléculaire avec le ruxolitinib était de 36mois versus non atteint dans le bras BAT.2
Le ropeginterféron alfa-2b réduit en continu la charge allélique
Le ropeginterféron alfa-2b est autorisé dans l’UE pour le traitement de première ligne de la MV depuis 2019. L’interféron (IFN) agit également par la voie de signalisation JAK-STAT et entraîne une diminution continue du fardeau allélique JAK2-V617F dans le traitement à long terme.7,8 Dans les études PROUD-PV et CONTINUATION-PV, l’IFN a été comparé à l’hydroxyurée (HU) pendant la première année de traitement et aux BAT (dont 88% de HU persistantes) à partir de la deuxième année. La réponse au ropeginterféron alfa-2b s’améliore continuellement avec la durée du traitement, avec une bonne tolérance des administrations sous-cutanées toutes les deux semaines.5,9 Dans l’évaluation finale des six années de suivi, non seulement la proportion de patient·es en rémission hématologique complète (RHC) durable était significativement plus élevée dans le bras interféron (72,6% vs 52,6%, p=0,004), mais l’EFS (thromboembolies, progression de la maladie ou décès) et la réponse moléculaire (69,1% vs 21,6%, p<0,0001) étaient également supérieures à celles obtenues avec les BAT.5,10 Environ la moitié des patient·es (50 sur 92) ont présenté une réduction de la charge allélique à <10% et 14 patient·es sur 92 présentaient une charge allélique <1% après 72mois de traitement par ropeginterféron alfa-2b.10
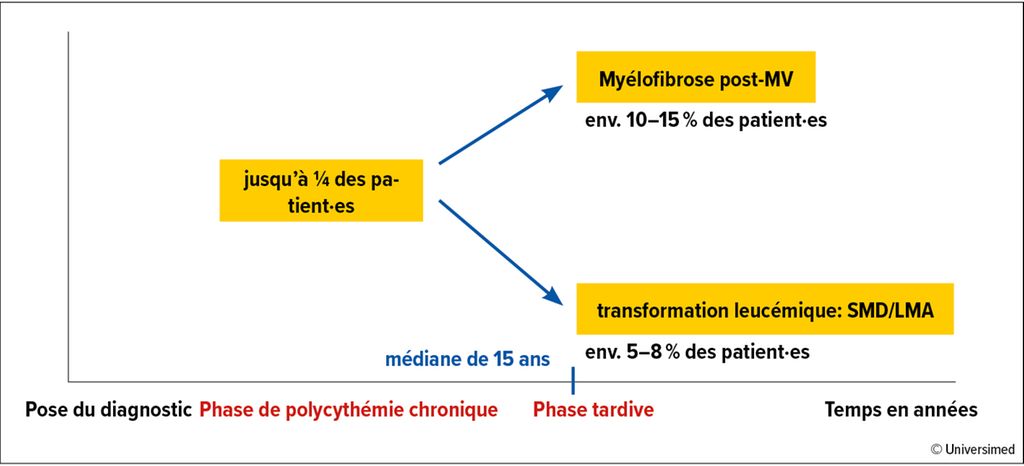
Fig.1: Risque de progression dans la MV
La diminution de la charge allélique de JAK2-V617F sert de marqueur de substitution pour la modification de la maladie, bien qu’il manque jusqu’à présent d’études prospectives évaluant dans quelle mesure une diminution de la charge allélique de JAK2 prévient la progression vers la myélofibrose ou un SMD/une LMA. En ce qui concerne la progression de la maladie, il convient de noter que chez plus de 50% des patient·es atteint·es de MV, des lésions sous-clonales non pilotes sont déjà détectables au moment du diagnostic, comme TET2 (sans signification pronostique), ASXL1 et LNK. Au cours de l’évolution, d’autres mutations typiques du SMD/de la LMA peuvent s’ajouter, comme les mutations SRSF2, RUNX1, TP53 ou IDH. Celles-ci sont prises en compte, en plus de l’âge, du nombre élevé de leucocytes et du caryotype anormal, dans le score MIPPS-PV («mutation-enhanced international prognostic score»). Les groupes de risques résultants «low», «intermediate-1», «intermediate-2» et «high» divisent la probabilité de survie en un spectre allant d’une médiane de 5,4ans pour le groupe à haut risque à une médiane de 25,3ans pour le groupe à bas risque.20
Trouver le traitement individuel optimal
Le défi consiste à mettre en place une thérapie sur mesure. Les symptômes souvent pénibles de la MV sont le prurit, les symptômes constitutionnels comme une fatigue prononcée, les troubles de la microcirculation (p.ex. érythromélalgie) et la survenue d’événements thromboemboliques malgré le traitement par AAS et la baisse de l’hématocrite en dessous de 45% conformément aux recommandations des lignes directrices. La classification classique en MV à faible risque et MV à haut risque se réfère à la probabilité de thromboembolies et ne comprend que deux facteurs: l’âge ≥60ans et la présence d’antécédents d’événements thromboemboliques. D’autres facteurs de thrombose et de progression de la maladie sont la leucocytose et une charge allélique JAK2-V617F élevée, mais ils n’entrent pas dans la classification des risques.16,17
Les saignées seules, associées à l’AAS ou à l’ajout d’hydroxyurée, ne permettent pas de contrôler de manière satisfaisante la charge symptomatique ni d’éviter les complications thromboemboliques à long terme. C’est pourquoi l’utilisation précoce de traitements modificateurs de la maladie, comme le ropeginterféron ou le ruxolitinib, fait l’objet d’une attention particulière.
Le ruxolitinib est autorisé en deuxième ligne depuis 2014 en cas de résistance ou d’intolérance à l’hydroxycarbamide.14 Cette substance est particulièrement adaptée au contrôle des symptômes et à la réduction de la taille de la rate.
Le ropeginterféron est autorisé en monothérapie dans la MV sans splénomégalie symptomatique, indépendamment du traitement antérieur ou de la présence de facteurs de risque d’événements thromboemboliques.15 L’utilisation du ropeginterféron est recommandée aussi bien chez les patient·es à haut risque que chez les patient·es à faible risque.11-13
Dans le registre espagnol de la maladie de Vaquez, 453patient·es à faible risque traité·es initialement uniquement par saignées et AAS ont été suivis pendant dix ans (2011–2021). Un contrôle durable de l’hématocrite n’a pu être obtenu que chez un tiers des patient·es et aucun contrôle satisfaisant des symptômes n’a été obtenu au cours de l’évolution. Les événements thrombotiques sont survenus à une fréquence de 0,8% par an (probabilité attendue de 8,5% sur dix ans). Une tendance à un risque supérieur de thrombose veineuse a été observée en cas de charge allélique JAK2 élevée.18 Sur la base de l’étude «Low PV», une étude randomisée de phaseII comparant le ropeginterféron alfa-2b et la phlébotomie, le ropeginterféron a également été approuvé pour le traitement de la MV à faible risque, avec pour objectif la modification précoce de la maladie.6,21
Le rôle de la saignée a évolué
Un problème de la thérapie par saignée seule en association avec l’AAS est également la valeur d’hématocrite fluctuante dans l’analyse longitudinale, car la saignée ne «coupe» toujours que ponctuellement la surcharge érythrocytaire. Cela ne permet pas d’obtenir une réponse hématologique continue. Dans une analyse rétrospective de données en situation réelle portant sur 28306patient·es atteint·es de MV (2011-2019), les patient·es du groupe à faible risque et du groupe à haut risque sous monothérapie par saignées ont présenté un contrôle suboptimal de l’hématocrite (défini comme Hk >50% parfois ou toujours). Cela concernait 54% des patient·es du groupe à haut risque et 64% du groupe à faible risque.22 Outre cette prise en charge insatisfaisante en termes de prévention des complications vasculaires et de réduction des symptômes spécifiques à la maladie, la prévention de la progression de la maladie n’est pas abordée dans la perspective à long terme.
Le rôle de la saignée a donc fondamentalement changé et réside dans la réduction initiale de l’hématocrite afin de prévenir les complications vasculaires aiguës. Pour le contrôle à long terme de la maladie, l’instauration précoce d’un traitement cytoréducteur est indiquée, avec une préférence pour le ropeginterféron alfa-2b.11,12
Objectifs thérapeutiques dans la maladie de Vaquez
Prévention des complications thromboemboliques à long terme
Réduction du risque de progression de la maladie vers une myélofibrose ou une leucémie aiguë
Amélioration de la qualité de vie par la réduction de la charge symptomatique
Éviter les saignées pour prévenir les symptômes de carence en fer
Littérature:
1 Baxter EJ et al.: Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 2005; 365(9464): 1054-61 2 Harrison CN et al.: Ruxolitinib versus best available therapy for polycythemia vera intolerant or resistant to hydroxycarbamide in a randomized trial. JCO 2023; 41(19): 3534-44 3 Kiladjian JJ et al.: Long-term efficacy and safety of ruxolitinib versus best available therapy in polycythaemia vera (RESPONSE): 5-year follow up of a phase 3 study. Lancet Haematol 2020; 7(3): e226-37 4 Barosi G et al.: Revised response criteria for polycythemia vera and essential thrombocythemia: an ELN and IWG-MRT consensus project. Blood 2013; 121(23): 4778-81 5 Gisslinger H et al.: Ropeginterferon alfa-2b versus standard therapy for polycythaemia vera (PROUD-PV and CONTINUATION-PV): a randomised, non-inferiority, phase 3 trial and its extension study. Lancet Haematol 2020; 7(3): e196-208 6 Barbui T et al.: Ropeginterferon alfa-2b versus phlebotomy in low-risk patients with polycythaemia vera (Low-PV study): a multicentre, randomised phase 2 trial. Lancet Haematol 2021; 8(3): e175-84 7 Abu-Zeinah G et al.: Interferon-alpha for treating polycythemia vera yields improved myelofibrosis-free and overall survival. Leukemia 2021; 35(9): 2592-601 8 Sørensen AL et al.: Ruxolitinib and interferon-α2 combination therapy for patients with polycythemia vera or myelofibrosis: a phase II study. Haematologica 2020; 105(9): 2262-72 9Kiladjian JJ et al.: Long-term outcomes of polycythemia vera patients treated with ropeginterferon alfa-2b. Leukemia 2022; 36(5): 1408-11 10 Gisslinger H et al.: Event free survival in patients with polycythemia vera treated with ropeginterferon alfa-2b versus best available treatment. Leukemia 2023; 37(10): 2129-32 11 Lengfelder E et al.: Onkopedia-Leitlinie Polycythaemia vera (PV), mise à jour de l’information septembre 2023 ( https://www.onkopedia.com/de/onkopedia/guidelines/polycythaemia-vera-pv/@@guideline/html/index.html ) 12 NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®) Myeloproliferative Neoplasms. Version 1.2024 ( https://www.nccn.org/professionals/physician_gls/pdf/mpn.pdf ) 13 Marchetti M et al.: Appropriate management of polycythaemia vera with cytoreductive drug therapy: European LeukemiaNet 2021 recommendations. Lancet Haematol 2022; 9(4): e301-11 14 Information professionnelle Jakavi, mise à jour de l’information avril 2022 15 Information professionnelle de Besremi, mise à jour de l’information février 2019 16 Vannucchi AM et al.: Prospective identification of high-risk polycythemia vera patients based on JAK2(V617F) allele burden. Leukemia 2007; 21(9): 1952-9 17 Carobbio A et al.: JAK2V617F allele burden and thrombosis: a direct comparison in essential thrombocythemia and polycythemia vera. Exp Hematol 2009; 37(9): 1016-21 18 Triguero A et al.: Low-risk polycythemia vera treated with phlebotomies: clinical characteristics, hematologic control and complications in 453 patients from the Spanish registry of polycythemia vera. Ann Hematol 2022; 101(10): 2231-9 19 Passamonti F et al.: Ruxolitinib versus best available therapy in inadequately controlled polycythaemia vera without splenomegaly (RESPONSE-2): 5-year follow up of a randomised, phase 3b study. Lancet Haematol 2022; 9(7): e480-92 20 Tefferi A et al.: Mutation-enhanced international prognostic systems for essential thrombocythaemia and polycythaemia vera. Br J Haematol 2020; 189(2): 291-302 21 Barbui T et al.: Ropeginterferon versus standard therapy for low-risk patients with polycythemia vera. NEJM Evid 2023; 2(6): EVIDoa2200335 22 Verstovsek S et al.: Real-world treatments and thrombotic events in polycythemia vera patients in the USA. Ann Hematol 2023; 102(3): 571-81
Das könnte Sie auch interessieren:

Sarcomes: des résultats prometteurs
Les sarcomes représentent une entité tumorale extraordinairement hétérogène et sont classés en plus de 100 sous-types selon l’OMS.1 Cela se reflète également dans les travaux de ...

AML au congrès de l’EHA: une grande déception et pourtant beaucoup d’espoir
La leucémie aiguë myéloïde a fait l’objet de nombreuses contributions lors du congrès 2024 de l’EHA. De nouvelles connaissances ont notamment été présentées sur la stratification du ...

Options sans chimiothérapie dans différents sous-groupes
Dans la prise en charge du cancer du sein avancé HR positif/HER2 négatif, on dispose désormais d’un paysage thérapeutique varié. Les approches ciblées, en plus de l’endocrinothérapie, ...